ГОМОГЕ́ННЫЙ КАТА́ЛИЗ
-

Рубрика: Химия
-

-

Скопировать библиографическую ссылку:
ГОМОГЕ́ННЫЙ КАТА́ЛИЗ, увеличение скорости химич. реакций, протекающих в газовой или жидкой фазе, в результате действия катализаторов, находящихся в одной фазе с реагентами. Гетерофазная реакция $\ce{СО+H2O <=> CO2 +H2}$ также может быть гомогенной каталитич. реакцией, поскольку проходит в объёме раствора катализатора (напр., $\ce{RhI3}$) с растворённым $\ce{CO}$.
Историческая справка
Впервые явление гомогенного газофазного катализа обнаружили в 1806 франц. химики Н. Клеман и Ш. Дезорм, установившие влияние оксидов азота на скорость окисления SО2 при произ-ве серной кислоты камерным (нитрозным) методом. Сознательное применение гомогенного катализа начинается с работы К. С. Кирхгофа по кислотному гидролизу крахмала до глюкозы (1811). Одним из первых шагов в развитии гомогенного металлокомплексного катализа можно считать открытие М. Г. Кучеровым в 1881 катализа солями ртути гидратации ацетилена. В 20 в. были открыты полимеризация ацетилена комплексами Cu(I) (амер. химик Ю. Ньюленд, рос. химик А. Л. Клебанский), гидроформилирование алкенов комплексами Co (нем. химик О. Рёлен), циклополимеризация ацетилена и карбонилирование ацетилена, алкенов и спиртов комплексами Ni(0) и Ni(II) (нем. химик В. Реппе), стереоспецифич. каталитич. полимеризация алкенов и диенов (К. Циглер, Дж. Натта – Нобелевская пр., 1963), катализ комплексами Pd(II) окисления алкенов до альдегидов и кетонов (в Германии – Ю. Смидт с сотрудниками, в России – И. И. Моисеев, М. Н. Варгафтик, Я. К. Сыркин), асимметрич. катализ гидрирования и эпоксидирования с применением хиральных комплексов Rh, Ru и Ti (У. Ноулз, Р. Нойори, Б. Шарплесс – Нобелевская пр., 2001), процессы метатезиса алкенов и метатезисной полимеризации циклоалкенов (И. Шовен, Р. Шрок, Р. Граббс – Нобелевская пр., 2005). Катализаторы на основе апротонных органич. сверхкислот были разработаны М. Е. Вольпиным с сотрудниками. Открытие процессов с участием комплексов металлов привело к созданию новой области каталитич. химии и пром. катализа – гомогенного металлокомплексного катализа. Важную роль в понимании сущности этого вида катализа как явления, связанного с превращениями молекул в координац. сфере металлокомплекса, сыграли работы И. И. Моисеева по изучению механизма реакций окисления алкенов в растворах комплексов Pd(II), Г. Стернберга, И. Уэндера, М. Орчина, Д. Бреслоу и Р. Хека (США) по исследованию механизма гидроформилирования алкенов в растворах комплексов Co(0), работы Дж. Хальперна (США) по изучению механизма активации Н2 комплексами металлов в реакциях восстановления неорганич. окислителей и гидрирования алкенов.
Характеристики гомогенно-каталитических процессов
Осн. характеристиками гомогенного каталитич. процесса являются величины активности катализатора и селективности катализируемой реакции. Селективность может быть представлена через долю прореагировавшего исходного реагента, превращённого в целевой продукт с учётом стехиометрии реакции. Для выражения каталитич. активности используют отношение начальной или стационарной скорости реакции к молярной концентрации активной формы катализатора – т. н. скорость (или частоту) оборотов катализатора (обозначается TOF, от англ. turn-over frequency). На практике часто применяют связанную с TOF, но не идентичную ей величину – отношение суммарного мольного количества продукта реакции к суммарному мольному количеству катализатора и ко времени реакции, которую также называют TOF. Наглядной характеристикой активности и стабильности работы катализатора является число оборотов катализатора (TON, turn-over number), равное числу каталитич. циклов в пересчёте на 1 моль катализатора (выражается отношением мольного количества продуктов реакции к мольному количеству катализатора).
Классификация гомогенно-каталитических процессов и их механизмы
Исходя из природы катализатора, т. е. специфич. возможностей для взаимодействия с субстратом, гомогенные каталитич. процессы подразделяют на следующие виды: кислотно-основный катализ протонными кислотами или основаниями Брёнстеда, электрофильный (с участием апротонных кислот Льюиса) и нуклеофильный (с участием оснований Льюиса) катализ, металлокомплексный катализ комплексными соединениями металлов, катализ органич. синтетич. соединениями, а также ферментативный катализ.
Кислотный катализ – активация субстратов, имеющих свободные электронные пары, протонными кислотами (см. Кислоты и основания) – происходит в результате присоединения протона кислоты НА к субстрату. Протонирование субстрата в водных растворах кислот является обычно реакцией замещения воды в гидратированном катионе $\ce{H(H2O)+}$ молекулой субстрата. Промежуточными активными частицами в кислотном катализе часто служат ионы карбения $\ce{R+}$, которые так же, как и протон, сольватированы молекулами $\ce{H2O}$, органич. растворителей или сильных кислот, напр. $\ce{R(H2O)+}$, $\ce{(C2H5)3O+}$, $\ce{RH2SO4+}$. Основный катализ – активация основаниями Брёнстеда – происходит в результате отщепления протона основанием от субстрата с образованием из молекулы субстрата анионной частицы, являющейся очень сильным нуклеофилом. Т. о., гидратацию алкенов в присутствии сильных минер. кислот – типичную кислотно-каталитич. реакцию – можно представить в виде последовательности стадий: $$\ce{RCH=CH2 +HA<=>RCH+CH3 +A- ,\\ RCH+CH3 +H2O<=>RCH(H2O)+CH3,\\ RCH(H2O)+CH3 +A- <=> RCH(OH)CH3 +HA};$$ альдольную конденсацию ацетона в присутствии щелочей – пример осно́вного катализа – в виде: $$\ce{CH3COCH3 +OH- <=> CH3COCH2- +H2O,\\ CH3COCH2- +CH3COCH3 <=> CH3COCH2C(O- )(CH3)2,\\ CH3COCH2C(O- )(CH3)2 +H2O <=> CH3COCH2C(OH)(CH3)2 +OH- .}$$
Очень сильные протонные кислоты (сверхкислоты) способны протонировать соединения, не имеющие свободных электронных пар, напр. алканы, с образованием ионов карбония $\ce{RH2+}$ ($\ce{CH5+}$ и др.). Ионы карбония участвуют в реакциях алкилирования, крекинга и изомеризации алканов.
Электрофильный катализ – активация электрофильными апротонными кислотами Льюиса – сопровождается понижением электронной плотности на реакционном центре субстрата (основания Льюиса) вплоть до образования иона карбения. По такому механизму происходит, в частности, алкилирование ароматич. соединений; напр., алкилирование бензола алкилбромидом по схеме $\ce{C6H6 + RBr -> C6H5R + HBr}$ включает образование реакционноспособного комплекса $\ce{R+[Al2Br7]-}$ в результате взаимодействия катализатора $\ce{Al2Br6}$ с алкилбромидом и действие катиона карбения $\ce{R+}$ на молекулу бензола.
В реакциях галогенсодержащих молекул ($\ce{CBr4, RCOCl, SO2Cl2}$ и др.) с $\ce{Al2Br6}$ или $\ce{Al2Cl6}$ возникают суперэлектрофильные частицы (например, $\ce{CBr3+Al2Br7-}$). Суперэлектрофилы катализируют крекинг алканов в мягких условиях.
Протонные и апротонные (электрофильные) катализаторы ускоряют процессы алкилирования, ацилирования, диеновый синтез и даже некоторые окислительно-восстановит. реакции. Напр., протонные кислоты катализируют окисление изопропанола трифенилкарбинолом до ацетона через стадию образования трифенилметильного катиона $\ce{(C6H5)3C+}$, апротонные кислоты (алкоголяты алюминия) – восстановление кетонов спиртами (Меервейна – Понндорфа – Верлея реакция) и диспропорционирование альдегидов (Тищенко реакция) через стадию образования комплекса между алкоголятом $\ce{Al}$ и карбонильным соединением.
Нуклеофильный катализ основаниями Льюиса происходит с образованием промежуточного продукта присоединения катализатора-нуклеофила к субстрату (напр., при электрофильном бромировании алкенов в присутствии галогенид-ионов) или с образованием промежуточного продукта замещения (примером замещения у насыщенного атома $\ce{C}$ является гидролиз алкилгалогенидов в присутствии аниона $\ce{I-}$ – активного нуклеофильного катализатора и затем легко замещаемой группы).
При катализе органич. соединениями функции катализаторов, как правило, более сложны, чем электрофилов или нуклеофилов. Примеры этого вида Г. к. – автокатализ гликолевым альдегидом конденсации формальдегида до сахаров в осно́вных средах (реакция Бутлерова), разложение пероксидных радикалов, катализируемое $n$-бензохиноном по схеме $$\ce{RR′C(OH)OO\!\bullet +n-C6H4O2 <=> RR′C=O +n-C6H4O2H\!\bullet +O2,\\ RR′C(OH)OO\!\bullet +n-C6H4O2H\bullet <=> RR′C(OH)OOH +n-C6H4O2},$$ катализ аминокислотой (пролином) альдольной конденсации, Манниха реакции и др. процессов.
В большинстве процессов катализ комплексами металлов реализуется через промежуточные металлокомплексные интермедиаты, в т. ч. и в типичных окислительно-восстановит. процессах с участием неорганич. реагентов. Напр., при катализе комплексами Мо(III) восстановления молекулярного азота амальгамой натрия по схеме $\ce{N2 + 4Na + 4H2O -> NH2NH2 +4NaOH}$ образующийся в результате взаимодействия $\ce{N2}$ с $\ce{Мо(III)}$ комплекс $\ce{[Mo^{4+} -N=N-Mo^{4+}]}$ при действии Na превращается в анион $\ce{[Mo^{4+}=N-N=Mo^{4+}]^{2–}}$; реакция этой промежуточной частицы с $\ce{H2O}$ и приводит к образованию гидразина (реакция открыта А. Е. Шиловым с сотрудниками). Лишь для небольшого числа реакций переноса электронов, катализируемых комплексами металлов, характерен происходящий без образования интермедиатов внешнесферный перенос электрона.
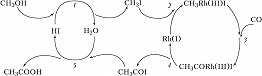
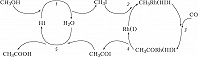
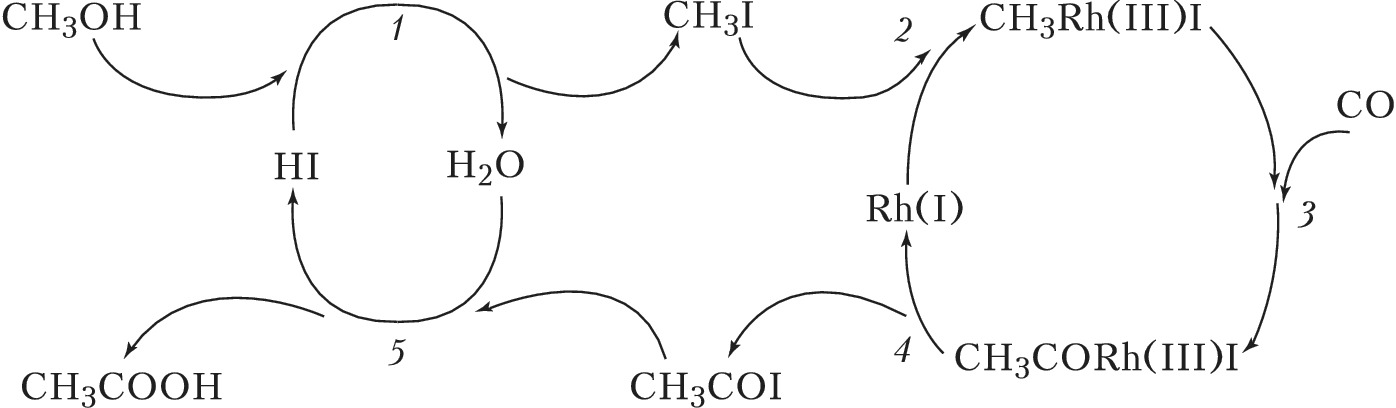
Наиболее распространённый тип металлокомплексного Г. к. – катализ реакций органич. соединений с образованием металлоорганич. интермедиатов со связями металл – углерод, т. н. металлоорганический катализ. Характерные стадии металлоорганич. катализа можно проиллюстрировать на примере двух процессов. Первый – пром. получение уксусной кислоты карбонилированием метанола в каталитич. системе $\ce{RhI3 – HI – H2O}$. Соль Rh(III) является прекурсором активного катализатора – комплекса $\ce{Rh(I)}$, образующегося по реакции $\ce{RhI3 + 3CO + H2O -> Rh(CO)2I2- + CO2 +HI +H+}$. Механизм процесса можно изобразить циклич. последовательностью стадий (рис. 1). Стадия 1 – замещение на галоген гидроксильной группы, стадия 2 – окислительное присоединение $\ce{CH3I}$ к $\ce{Rh(I)}$, стадия 3 – внедрение $\ce{CO}$ по связи $\ce{CH3 -Rh}$, стадия 4 – восстановительное элиминирование ацилиодида $\ce{CH3COI}$, стадия 5 – нуклеофильное замещение $\ce{I-}$ в ацилиодиде водой. В этом процессе, кроме комплекса $\ce{Rh(I)}$, участвует протонный кислотный катализатор $\ce{HI}$ в двух каталитич. циклах. Такие системы называют полифункциональными каталитич. системами.
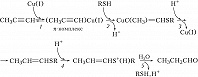
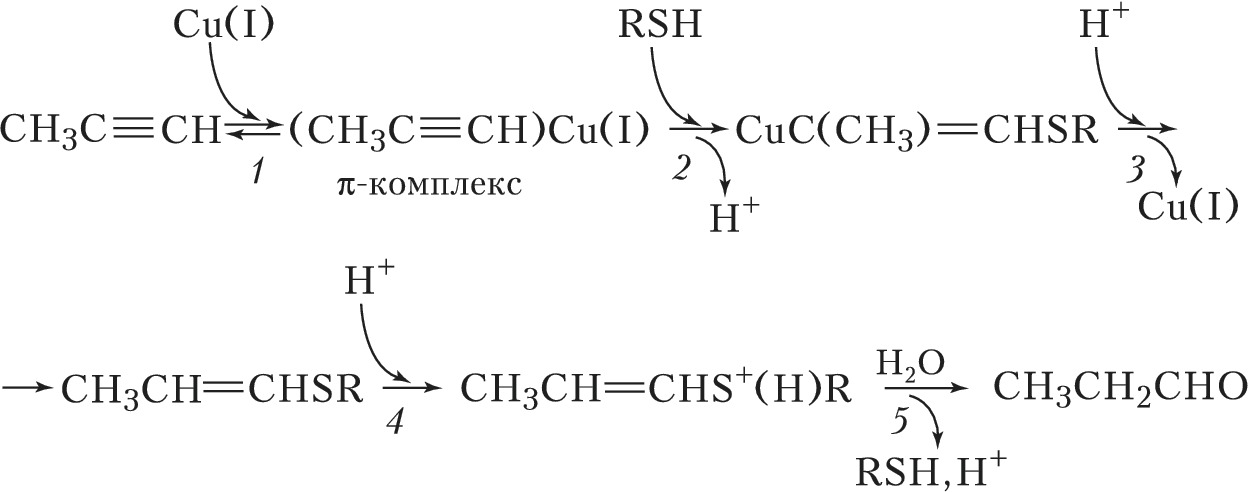
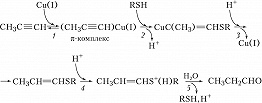
Второй пример – гидратация алкинов с участием трёх катализаторов: комплексов $\ce{Cu(I)}$ (металлокомплексный катализ), тиола $\ce{RSH}$ (нуклеофильный катализ) и $\ce{HCl}$ (протонный кислотный катализ), протекающая против правила Марковникова (рис. 2). Стадия 1 – образование $π$-комплекса, стадия 2 – нуклеофильное присоединение RSH к $π$-комплексу, стадия 3 – электрофильное замещение $\ce{Cu(I)}$ протоном, стадия 4 – электрофильное присоединение $\ce{H+}$ (протонирование тиопропенилового эфира), стадия 5 – нуклеофильное замещение тиола водой.
В металлокомплексном катализе выделяют асимметрический катализ с использованием хиральных металлокомплексных катализаторов, позволяющих проводить реакции стереоселективно (см. в ст. Асимметрический синтез). Напр., в пром-сти на комплексах $\ce{Rh(I)}$ с хиральными фосфиновыми лигандами получают дигидроксифенилаланин (препарат для лечения болезни Паркинсона).
Важная технологич. проблема металлокомплексного катализа – отделение катализаторов от продуктов и рециклизация катализаторов – решается за счёт иммобилизации комплексов металлов с помощью лигандов на поверхности носителей или в одной из фаз при использовании двухфазных систем (напр., органич. фаза и вода, в которой растворён комплекс металла), использования расплавов органич. солей (ионных жидкостей), в которых иммобилизован комплекс металла, применения мембран для отделения продуктов ультрафильтрацией, а также использования термоморфных лигандов или растворителей, меняющих в зависимости от темп-ры фазовое состояние.
Практическое применение
К числу важнейших пром. гомогенно-каталитич. процессов (кроме названных выше) относятся синтезы с участием CO, олигомеризация этилена с кросс-метатезисом терминальных и интернальных алкенов, димеризация этилена и пропилена, гидрирование функционально замещённых алкенов, нитросоединений, эпоксидирование пропилена, окисление алкилароматич. соединений и др. Мн. металлокомплексные каталитич. процессы по активности катализаторов, хемо-, регио- и стереоселективности приближаются к ферментативным. Использование структурных и функциональных моделей ферментов, принципов протекания биохимич. процессов позволяет создавать эффективные процессы металлокомплексного катализа (см. Биомиметические реакции).