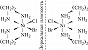
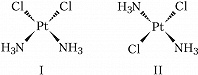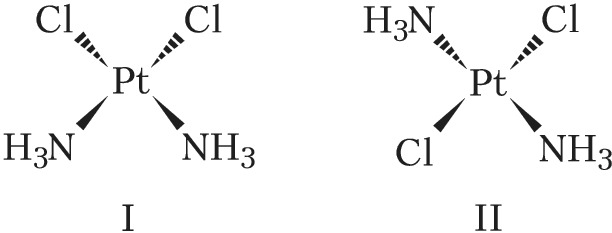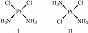КО́МПЛЕКСНЫЕ СОЕДИНЕ́НИЯ
-

Рубрика: Химия
-

-

Скопировать библиографическую ссылку:
КО́МПЛЕКСНЫЕ СОЕДИНЕ́НИЯ (координационные соединения), химические соединения, в которых можно выделить центр. атом (комплексообразователь) и непосредственно связанные с ним (координированные) один или неск. ионов и/или молекул. Координированные частицы называются лигандами, число донорных атомов в них, координированных центр. атомом, – его координационным числом. Центр. атом связывает лиганды как за счёт электростатического, так и за счёт донорно-акцепторного взаимодействия. Координационное число и степень окисления являются важнейшими характеристиками атома-комплексообразователя.
Центр. атом и координированные лиганды образуют внутр. координационную сферу К. с.; при написании формулы К. с. её обычно заключают в квадратные скобки. Внутри скобок запись производится в следующей последовательности: химич. символ центр. атома, символы анионных, затем нейтральных лигандов с указанием их числа. Если внутр. сфера несёт заряд, то его компенсируют противоионы, образующие внешнюю сферу. Внешнесферными могут быть и катионы, напр. $\ce{K^+}$ в $\ce{K_4[Fe(CN)_6]}$, и анионы, напр. $\ce{SO^{2-}_4}$ в $\ce{[Сu(NH_3)_4]SO_4}$. Кроме противоионов, во внешней сфере могут находиться нейтральные молекулы. Примерами К. с., состоящих только из центр. атома и лигандов, могут служить $\ce{Ti(CO)7}$, $\ce{Cr(CO)6}$ и др. карбонилы металлов.
Названия К. с. строятся в соответствии с номенклатурными правилами ИЮПАК начиная с лигандов и учитывая их заряд; напр., $\ce{[PtCl_2(NH_3)_2]}$ – дихлородиамминплатина(II), $\ce{[Co(NH_3)_6](NO_3)_3}$ – нитрат гексаамминкобальта(III), $\ce{Na_2[PdCl_4]}$ – тетрахлоропалладат(II) натрия.
Историческая справка
Среди ранних, научно документированных, исследований К. с. можно выделить получение $\ce{[Cu(NH_3)_4]Cl_2}$ в 1597 А. Либавием и $\ce{KFe[Fe(CN)_6]}$ в 1704 нем. ремесленником Г. Дисбахом, однако в соответствии с существующими на тот период представлениями эти вещества относили к двойным солям. Началом систематич. изучения К. с. обычно считают открытие франц. химика Б. Тассера, описавшего в 1798 появление коричневой окраски в аммиачных растворах хлорида кобальта при образовании хлорида гексаамминкобальта(III) $\ce{[Co(NH_3)_6]Cl_3}$. Важной особенностью этого исследования было понимание того, что образующееся соединение является продуктом сочетания способных к самостоят. существованию валентно-насыщенных, весьма устойчивых «простых» соединений и что для водных растворов образующегося «сложного», или комплексного (от лат. complexus – сочетание), соединения характерны свойства, отличные от свойств составляющих его простых соединений. В 19 в. было синтезировано большое число разнообразных К. с.; среди эксперим. исследований можно выделить работы дат. химиков В. Цейзе, получившего К. с. платины с органич. лигандами $\ce{K[PtCl_3(C_2H_4)]}$ (соль Цейзе, 1827), и С. Йёргенсена (синтезировал К. с. кобальта, хрома, родия, платины). В этот же период Т. Грэм, К. К. Клаус и др. учёные делают попытки объяснить существование и структуру К. с. Из ранних теорий наиболее широко известна цепная теория швед. химика К. Бломстранда, развитая С. Йёргенсеном (теория Бломстранда – Йёргенсена, 1869), позволившая объяснить строение некоторых классов К. с. (в частности, аммиакатов). Обобщённое представление о пространственном строении К. с. дала координационная теория, предложенная А. Вернером в 1893 (работа удостоена в 1913 Нобелевской пр.). Координационная теория опровергала общепринятые для объяснения строения неорганич. соединений представления о постоянной и направленной валентности. А. Вернер ввёл важные для целого историч. периода понятия «главной» и «побочной» валентности, координации, координационного числа, геометрии К. с., создал основы классификации К. с.; вопрос о природе главной и побочной валентности в координационной теории не рассматривался. Разрешение вопроса о природе координационной связи стало возможным после создания электронной теории валентности (Г. Льюис, 1916). Осн. заслуга в использовании этой теории для объяснения природы координационной связи принадлежит англ. химику Н. Сиджвику. Согласно концепции Сиджвика (1923), главные валентности были интерпретированы как результат переноса электрона, побочные – как результат обобществления электронных пар. Развитие совр. представлений о природе координационной связи связано с использованием квантовохимич. подходов – теории кристаллич. поля, метода валентных связей, метода молекулярных орбиталей, теории поля лигандов; осн. вклад в распространение на К. с. метода валентных связей принадлежит Л. Полингу, теории кристаллич. поля – амер. химику Л. Оргелу. Развитию химии К. с. способствовали исследования амер. учёных Дж. Бейлара, Р. Пирсона, Г. Грея, отеч. химиков И. И. Черняева, Л. А. Чугаева, А. А. Гринберга, К. Б. Яцимирского и др.
В течение длительного периода химия К. с. – координационная химия – считалась одним из разделов неорганич. химии, поскольку большинство известных К. с. содержало в качестве лигандов неорганич. молекулы или ионы (аммиак, воду, цианогруппу и т. п.). Выделение координационной химии в самостоятельный, интенсивно развивающийся раздел химич. науки связано не только с многочисленностью К. с. (К. с. по распространённости – вторые после органич. соединений, известны практически для всех элементов-металлов и для некоторых неметаллов, содержат как неорганические, так и органич. лиганды самых разнообразных типов), стремительным ростом количества изучаемых объектов и открытием новых классов К. с. (металлоорганических соединений $\pi$-комплексного типа, природных К. с. и их синтетич. аналогов и пр.), но и с разработкой теоретич. представлений, позволяющих рассматривать разнообразные классы К. с. на единой основе. Междисциплинарное положение координационной химии обусловливает необходимость использования для её развития методов неорганической, физической, органической, аналитической, структурной химии. Изучение химич. и физико-химич. свойств К. с. способствует установлению закономерностей, представляющих интерес для органической, биологич. химии, катализа, электрохимии, фотохимии, химич. технологии, материаловедения, медицины и др. смежных областей.
Классификация комплексных соединений
Сложность классификации К. с. обусловлена их многообразием. Наиболее общие принципы классификации К. с. следующие: 1) по заряду: нейтральные, напр. $\ce{[PtCl_2(NH_3)_2]}$; катионные, напр. $\ce{[Cr(NH_3)_6]Cl_3}$; анионные, напр. ацидокомплексы (лигандами служат анионы кислот – ацидогруппы) – $\ce{K_4[Fe(CN)_6], K[AuCl_4]}$ и др.; катионно-анионные, напр. $\ce{[Cr(NH_3)_6][Fe(CN)_6]}$; молекулярные, напр.$\ce{Ni(CO)_4}$; 2) по типу лигандов: простые, содержащие монодентатные лиганды, напр. $\ce{[Cu(NH_3)_4]Cl_2}$; хелатные – с присоединёнными к одному центр. атому через два или более соединительных, координирующих атома хелатными лигандами, напр.$\ce{[Cu(en)_2]Cl_2}$ (en – этилендиамин $\ce{H_2NCH_2CH_2NH_2}$ имеет два координирующих атома азота); содержащие лиганды одного типа, напр. $\ce{[Co(NH_3)_6]Cl_3}$; содержащие разл. лиганды, напр. $\ce{[Co(NH_3)_4Cl_2]}$; 3) по количеству атомов элемента-комплексообразователя: моноядерные (все вышеприведённые примеры); полиядерные (или многоядерные), напр. $\ce{[(en)_2Cr(OH)_2Cr(en)_2]Br_4}$; к полиядерным К. с. относятся также кластеры, металлоцены, комплексы с мостиковыми лигандами и некоторые др. соединения. К комплексам с мостиковыми лигандами относятся гетерополисоединения – К. с. анионного типа, содержащие во внутр. сфере в качестве лигандов анионы неорганич. изополикислот (молибденовых, вольфрамовых и др.); изополианионы содержат мостиковые связи $\ce{М-О-М}$, где $\ce{М}$ – атом-комплексообразователь $\ce{(P, As, Si, Ge, Ti, Ce)}$, напр. $\ce{K_3[PMo_{12}O_{40}], K_8[Co_2W_{12}O_{42}]}$.
В отд. группы выделяют К. с. с одинаковыми лигандами: аквакомплексы (лигандами служат молекулы воды $\ce{H_2O}$), напр. $\ce{[Co(H_2O)_6]Cl_2}$; аммины (лиганды – молекулы аммиака $\ce{NH_3}$), напр. $\ce{[Pt(NH_3)_4]Cl_2}$, в эту же группу входят аммиакаты – К. с., содержащие молекулы аммиака не только во внутренней, но и во внешней сфере; гидроксокомплексы (лиганды – гидроксид-ионы $\ce{OH^{–}}$), напр. $\ce{K_2[Rb(OH)_6]}$; гидридные комплексы (лиганды – гидрид-ионы $\ce{Н^{–}}$), напр. $\ce{Na[AlH_4], Li[BH_4]}$; галогенаты (содержат атом галогена в качестве комплексообразователя и галогенидные лиганды); некоторые др. Галогенаты, в свою очередь, подразделяются на анионгалогенаты, напр. $\ce{Rb[Br(Br)_2], NH_4[ICl_4]}$ (соответственно изополигалогенат и гетерополигалогенат), и катионгалогенаты, напр.$\ce{[BrF_2][AsF_6]},$$\ce{[ICl_2][SbCl_6]}.$
Строение комплексных соединений
Химич. связь в К. с. – координационная связь – осуществляется либо за счёт размещения неподелённой электронной пары донорного атома лиганда на свободных (и доступных) электронных орбиталях центр. атома (акцептора), либо за счёт перехода собственных электронов металла-комплексообразователя на незаполненные орбитали лиганда. В последнем случае чаще всего это молекулярные разрыхляющие $\pi$-орбитали, поэтому такая связь называется $\pi$-донорной, или $\pi$-дативной. Наиболее наглядную качественную информацию об образовании координационной связи даёт метод валентных связей. Детальные теоретич. представления о строении К. с. отражены в методе молекулярных орбиталей, теории кристаллич. поля и теории поля лигандов. В рамках этих подходов даются объяснения электронного и геометрич. строения К. с., проводятся оценки энергии связей. В совр. теориях строения К. с. и природы координационной связи используются представления о кислотах и основаниях Льюиса, принцип Пирсона о мягких и жёстких кислотах и основаниях (см. в ст. Кислоты и основания).
Центр. атомом в К. с. может быть как металл, так и неметалл. Прочность координационной связи металл – лиганд тем выше, чем выше заряд иона-комплексообразователя и чем меньше его радиус. Существенную роль играет электронная структура центр. атома. Ионы с электронной конфигурацией инертного газа обладают наименьшей склонностью к комплексообразованию. Более сильными комплексообразователями являются ионы 3d-элементов, имеющие как незавершённые, так и завершённые электронные оболочки. Из-за большего радиуса и размытости электронных орбиталей ионы 4d-, 5d-, 4f-элементов и особенно 5f-элементов образуют менее прочные связи. Эти общие закономерности обусловлены характером заполненности электронной оболочки металла, а также стерическими требованиями – оптимальным соотношением между размерами центр. атома и лигандов. В качестве атома-комплексообразователя чаще всего выступают атомы переходных металлов ($\ce{Ti, V, Cr, Mn, Fe, Co, Ni, Сu, Zn, Zr, Nb, Mo, Ru, Rh, Pd, Ag, Cd, Hf, Ta, W, Re, Os, Ir, Pt, Au, Hg}$, редкоземельных элементов, актиноидов). Из неметаллов в качестве центр. атомов чаще всего выступают атомы $\ce{B, P, Si}$.
Лигандами в К. с. могут быть анионы неорганич. и органич. кислот ($\ce{F^{–}, Cl^{–}, Br^{–}, I^{–}, CN^{–}, NO2^{-} , SO4^2^{-} , PO4^3^{-} , C2O4^2^{-}}$ и др.), разл. нейтральные молекулы, ионы и свободные радикалы, содержащие атомы $\ce{O, N, P, S, Se, C}$. Активность лиганда зависит от природы донорного атома: жёсткие катионы (щелочных, щёлочноземельных металлов, лантаноидов) предпочтительно связываются донорным атомом кислорода, более мягкие (переходных d-элементов) – донорными атомами $\ce{N, S}$ и др. Лиганд с несколькими донорными атомами (напр., ЭДТА) способен образовывать хелатные циклы, обладающие высокой устойчивостью. Существенную роль при образовании К. с. играет строение (в т. ч. гибкость) молекулы лиганда. По своей способности внедряться в электронные оболочки центр. атома, приводящей к изменению её строения, лиганды условно подразделяют на лиганды сильного и слабого поля.
При образовании К. с. металл-комплексообразователь предоставляет свои валентные электронные орбитали (как заполненные, так и свободные) для размещения на них донорных электронных пар лигандов. Число и направленность заполненных общими электронами орбиталей определяют пространственное строение – стереохимию – К. с. Так, $sp$-комбинация молекулярных орбиталей соответствует линейному строению К. с., напр. $\ce{[Ag(NH_3)_2]^+}$; $sp^2$ – плоскому треугольному, напр. $\ce{(AlF_3)}$; $sp^3$ – тетраэдрическому, напр.$\ce{(Zn(NH_3)_4]^{2^{+}}}$; $sp^3d$ – тригонально-бипирамидальному, напр. $\ce{(NbF_5)}$; $dsp^2$ – плоскому квадратному, напр. $\ce{[Ni(CN)_4]^{2^{–}}}$; $d^2sp^3$ или $sp^3d^2$ – октаэдрическому, напр.$\ce{[Co(NH_3)_6]^{3^{+}}}$, и т. д. Пространственное расположение лигандов вокруг центр. атома характеризуется координационным полиэдром.
Изомерия комплексных соединений
Многообразие К. с. обусловлено образованием изомеров, одинаковых по составу, но отличающихся расположением лигандов вокруг центр. атома.
Гидратная (сольватная) изомерия обусловлена разл. распределением молекул воды и анионных лигандов между внутренней и внешней сферами К. с. Напр., соединение $\ce{CrCl_3·6H2O}$ существует по крайней мере в трёх изомерных формах: $\ce{[Cr(H_2O)_6]Cl_3}$ – трихлорид гексааквахрома(III) фиолетового цвета, $\ce{[CrCl(H_2O)_5]Cl_2·H_2O}$ – моногидрат дихлорид пентааквахлорохрома(III) сине-зелёного цвета и $\ce{[CrCl_2(H_2O)_4]Cl· 2H_2O}$ – дигидрат хлорид тетрааквадихлорохрома(III) зелёного цвета. Эти изомеры по-разному реагируют с раствором $\ce{AgNO_3}$, поскольку в осадок ($\ce{AgCl}$) переходит только хлор, содержащийся во внешней сфере.
Ионизационная изомерия характеризуется разл. распределением ионов между внешней и внутр. сферами К. с. При диссоциации в растворе такие изомеры образуют разные ионы. Напр., для соединения $\ce{CoBrSO_4·5NH_3}$ известны два изомера: $\ce{[CoBr(NH_3)_5]SO_4}$– красно-фиолетового цвета и $\ce{[CoSO_4(NH_3)_5]Br}$ – красного цвета.
Координационная изомерия заключается в различном распределении лигандов во внутр. координационных сферах. Напр., изомеры$\ce{[Co(NH_3)_6][Cr(CN)_6]}$ и $\ce{[Cr(NH_3)_6][Co(CN)_6]}$ по-разному взаимодействуют с $\ce{AgNO_3}$ : $\ce{[Co(NH_3)_6][Cr(CN)_6]}$ образует осадок состава $\ce{Ag_3[Cr(CN)_6]}$, $\ce{[Cr(NH_3)_6][Co(CN)_6]}$ приводит к осаждению соединения $\ce{Ag_3[Co(CN)_6]}$.
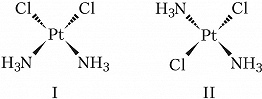
Геометрич. изомерия (цис, транс-изомерия) обусловлена различным пространственным расположением лигандов вокруг центр. атома. Так, комплекс $\ce{[PtCl_2(NH_3)_2]}$ существует в виде цис-изомера (формула I) и транс-изомера (формула II), отличающихся друг от друга рядом свойств.
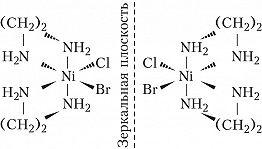
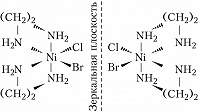
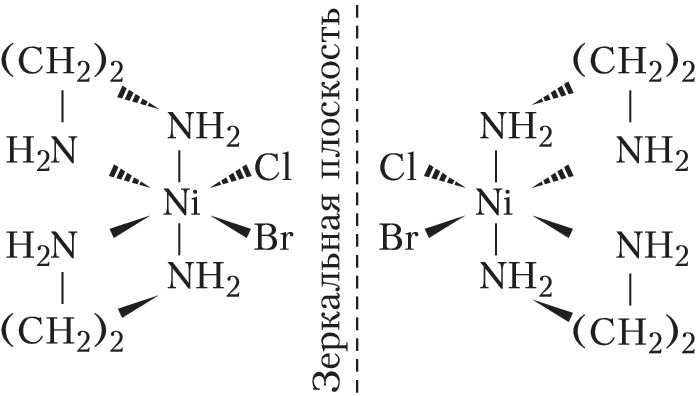
Оптич. изомерия характеризуется способностью вращать плоскость поляризации плоскополяризованного света. Два изомера – правый и левый – отличаются друг от друга направлением вращения. Эти изомеры – зеркальные изображения друг друга – не могут быть совмещены в пространстве. Из двух геометрич. изомеров бис-(этилендиамин)бромохлороникеля(II) $\ce{[NiClBr(en)_2]}$ только цис-изомер может существовать в виде двух оптич. изомеров – энантиомеров:
Структурными (конформационными) изомерами называют такие координационные изомеры, в которых происходит изменение симметрии (пространственного строения) координационной сферы.
Свойства комплексных соединений
Различают термодинамич. стабильность К. с. – меру возможности образования К. с. или его превращения в др. соединение в равновесных условиях, и кинетическую, описывающую скорость реакций комплексов, ведущих к достижению равновесия. Термодинамич. стабильность К. с. характеризуется терминами «устойчивый», «неустойчивый», кинетическая – «лабильный» и «инертный». Если при комнатной темп-ре реакция комплекса протекает за время смешения реагентов (ок. 1 мин), К. с. относят к лабильным; если реакция протекает с измеримой скоростью и половина времени жизни комплекса более 2 мин, такие К. с. относят к инертным. Напр., константа скорости изотопного обмена молекул воды во внутр. координационной сфере для инертного комплекса $\ce{[Ni(H_2O)_6]^{2^{+}}}$ равна $3,3· 10^4 с^{–1}$, для лабильного $\ce{[Сr(Н_2О)_6]^{3^{+}} – 5·10^{–7} c^{–1}}$.
Устойчивость К. с. определяется природой центр. атома и лиганда, а также стерическими факторами. В соответствии с принципом жёстких и мягких кислот и оснований, все центр. атомы условно разделяют на жёсткие и мягкие кислоты Льюиса. Жёсткие кислоты Льюиса имеют малый атомный или ионный радиус, высокую положительную степень окисления, предпочтительно взаимодействуют с неполяризующимися жёсткими основаниями, такими как $\ce{F^{–}, OH^{–}, NR2^{–}}$ ($\ce{R}$ – органич. радикал). К жёстким кислотам Льюиса относятся ионы элементов в высших степенях окисления с электронной конфигурацией $d^0$ или $d^{10}$. Мягкие кислоты Льюиса имеют большой атомный или ионный радиус и низкую степень окисления, более эффективно взаимодействуют с легко поляризующимися мягкими лигандами, такими как $\ce{SR2, PR3, I^{–}}$, олефины. Мягкие кислоты Льюиса имеют электроны на $d$-орбиталях, способные к образованию $\pi$-связей в результате перекрывания с вакантными $d$-орбиталями мягких лигандов. Эти же центр. ионы образуют К. с. с олефинами (типа соли Цейзе). Поскольку реакции комплексообразования подразумевают взаимодействие кислот и оснований Льюиса, с увеличением оснóвных свойств лигандов устойчивость комплексов повышается. Лиганды с большей основностью при введении в раствор полностью замещают во внутр. сфере лиганды с меньшей основностью.
Количественной характеристикой устойчивости К. с. служит его константа устойчивости $K=\ce{[ML_n]/([M][L]^{n}}) }$, где $\ce {[ML_{n}], [M], [L] }$ – равновесные концентрации комплекса, комплексообразователя и лиганда соответственно. Для эксперим. определения константы устойчивости применяют физико-химич. методы, позволяющие рассчитать равновесные концентрации ($рН$-метрическое титрование, кондуктометрию, спектрофотометрию, ЯМР-спектроскопию, полярографию, вольтамперометрию и др.).
Свободная энергия Гиббса реакции образования комплекса $ΔG^0$ связана с $K$, энтальпийным вкладом ($ΔH^0$) и энтропийным вкладом ($ΔS$) соотношением: $–RT\text{ln}K=ΔG^0=ΔH^0-TΔS^0$, где $T$ – абсолютная темп-ра, $R $ – газовая постоянная. В реакциях комплексообразования энтальпийный вклад обусловлен разностью в суммарной энергии связей исходных частиц и образующегося К. с.; обычно величины $ΔH$ невелики. Энтропийный вклад связан с изменением числа частиц в реакции. Потеря подвижности иона металла и лигандов при соединении их в К. с. обычно компенсируется за счёт высвобождения большого количества молекул растворителя (воды) из сольватных (гидратных) оболочек центр. атома и лигандов. Об устойчивости хелатных К. с. см. в ст. Хелаты.
К. с. участвуют в реакциях присоединения, замещения или элиминирования лиганда, реакциях изомеризации координационного полиэдра, реакциях связанного лиганда (напр., диссоциация, изомеризация лиганда) и реакциях электронного переноса.
Методы синтеза комплексных соединений
В молекуле К. с. можно сочетать разл. металлы и лиганды, что позволяет варьировать состав К. с., их строение и свойства. Используя соответствующую методику синтеза, можно получить К. с. с практически любыми заданными свойствами и в любом агрегатном состоянии. Многочисл. методы синтеза К. с. можно классифицировать по типам реакций, лежащих в их основе (реакции замещения, обмена, окислительно-восстановительные и т. д.). Выбор методики синтеза зависит от природы К. с. (термодинамич. устойчивости, кинетич. инертности или лабильности) и в соответствии с этим основывается на термодинамических или на кинетических подходах. К группе методов, основанных на термодинамич. подходе, относятся реакции, направление которых определяется термодинамич. факторами: энергетич. выгодностью образования продукта реакции по отношению к исходным соединениям (отрицательным изменением энергии Гиббса). В этих методах механизм реакций не играет существенной роли в процессе синтеза. В методах, основанных на кинетич. подходе, строение продукта генеалогически связано с исходными соединениями, синтез протекает в осн. с использованием реакций замещения и важную роль играет их механизм. В этом случае образование продукта может быть энергетически выгодно, но возможно и получение метастабильных К. с., образование которых энергетически менее выгодно по сравнению с др. продуктами.
Специфич. методом получения К. с. является темплатный синтез, когда сложные органич. лиганды образуются в процессе взаимодействия иона металла с более простыми органич. соединениями. Ион металла – матрица, на которой закрепляются исходные лиганды, – способствует пространственной ориентации лигандов и тем самым определяет направление реакции их взаимодействия. При отсутствии ионов металла-комплексообразователя реакция не протекает или протекает с малым выходом. Темплатный синтез наиболее эффективен для получения макроциклич. лигандов.
Области применения комплексных соединений
Металлоорганич. К. с. – один из наиболее перспективных классов химич. соединений, на основе которых могут быть созданы молекулярные материалы. Сочетание в одной молекуле ионов металлов и органич. лигандов, возможность целенаправленного изменения состава и строения К. с. открывают возможности для создания на их основе молекулярных материалов с широким диапазоном функциональных свойств – оптических, магнитных, электрических и т. д. К. с. применяют для выделения и очистки платиновых металлов, золота, серебра, никеля, кобальта, меди, в процессах разделения редкоземельных элементов, щелочных металлов и в ряде др. технологич. процессов. К. с. используют в химич. анализе для качественного обнаружения и количественного определения мн. химич. элементов. В живых организмах разл. типы К. с. представлены соединениями ионов металлов ($\ce{Fe, Cu, Mg, Mn, Mo, Zn, Co}$) с белками (металлопротеиды), витаминами, коферментами, др. веществами, выполняющими специфич. функции в обмене веществ. Природные К. с. участвуют в процессах дыхания, фотосинтеза, биологич. окисления, в ферментативных процессах.
К. с. используют в экстракционных и сорбционных процессах разделения и тонкой очистки редких, цветных и благородных металлов, в аналитич. химии. К. с. применяют в качестве селективных катализаторов разл. процессов химич. и микробиологич. пром-сти, для создания окислителей на основе фторидов галогенов и благородных газов, в качестве источников H2 и O2 на основе гидридов и кислородсодержащих соединений, в медицине, в т. ч. в терапии разл. видов опухолей, в качестве источника микроэлементов в животноводстве и с. х-ве, для получения тонких покрытий на разл. изделиях микроэлектроники для придания антикоррозионных свойств и механич. прочности.